|
Our main research in computational biology is on
interaction of RNA molecules.
The
goal is to find an optimal structure resulting from the folding of
the individual RNAs on the one hand, and the simultaneous binding of the
RNAs on the other hand, resulting in a RNA complex. The traditional
approach was to concatenate all RNAs into one and use folding
to simulate the interaction. This approach has limitations; for
instance, it cannot produce a structural form known as a kissing loop.
Our original formulation of the problem was for two RNAs, which led to
the introduction of RNA-RNA interaction graphs.
In these graphs, the optimal structure is given by a set of non-intersecting
edges of maximum weight, which is NP-hard. We have a 2/3 factor
approximation algorithm for this problem.
Our notion of
an "entangler" (recursive entanglers shown on the side)
in the RNA-RNA interaction
graph is instrumental in characterizing the sub-optimality of the predicted RNA complexes.
We proved that every
entangler-free solution for the RNA interaction problem is at most a
2/3 factor approximation of the optimal solution. The significance of
this result lies in the fact that the wide-spread dynamic programming
algorithms for the problem cannot produce entanglers, and thus are at
most 2/3 optimal.
We recently pioneered a
formulation for the multiple (two or
more) RNA interaction problem.
Multiple RNA interaction poses new
computational problems which also fall under the realm of combinatorial
optimization and NP-hardness. We developed approximation algorithms and, more
generally, algorithms that belong to a class known as PTAS
(Polynomial Time Approximation Scheme) in
computer science.
In a recent development, we illustrated how the formulation can
lend itself to find sub-optimal solutions efficiently. This is highly
significant because it helps identify relevant biological structures
that are not necessarily optimal in the computational sense.
We explored randomized algorithms in conjunction with
Gibbs sampling and MCMC (Monte Carlo Markov Chain). To appreciate the
challenges of this approach, observe that
many artifact interactions can easily arise when the RNAs are exact
complement of each other (they bind perfectly). The CopA-CopT complex
represents such an example. It is known as the perfect
couple, and has been problematic since the inception of pairwise
interaction algorithms in 2005. The correct solution
must drop many of these artifacts and, therefore, is typically very far from
optimal and will be missed by sampling. We developed a new scoring
scheme where sub-optimal solutions are not too detrimental, and hence
can still show up as candidates for the sampling algorithms.
We continue to work on the multiple RNA interaction problem.
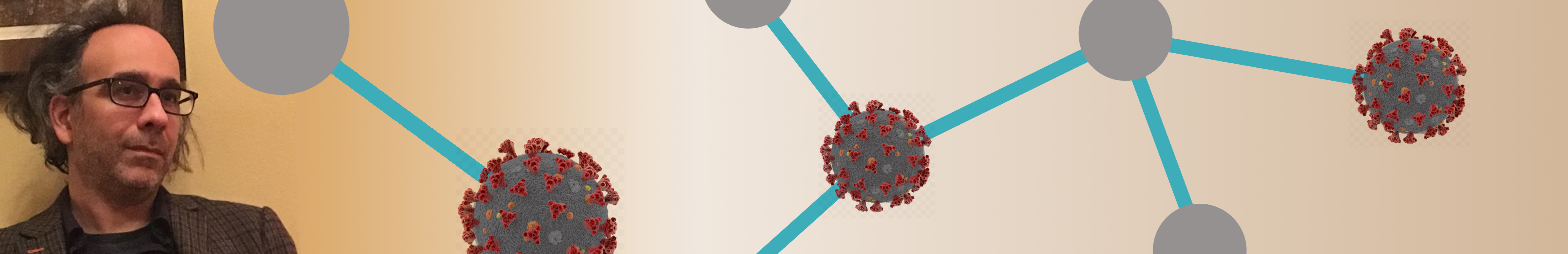
The current formulation
allows RNAs to interact on a path; for instance,
RNA 1 interacts with RNA
2, RNA 2 interacts with RNA 3, and RNA 3 interacts with RNA 4 (as shown on the side). More
recently, we extended this formulation to cycles, where in the above
example RNA 4 can also interact with RNA 1. While a path or a cycle is
sufficient for most RNA interactions, the order of the RNAs on the
path can dramatically alter the solution. In addition, this limits
each RNA to interact with at most two others. The goal is to generalize
the formulation to allow paths, cycles, trees, and other types of
graphs. The general problem has relations to other problems in computer science such as increasing sequences,
block designs, graph arboricity, and graph decompositions.
|



|